In order for the semiconductor industry to continually stay on Moore’s Law, it is required that the wavelength of exposure tools must shrink over time. At these extremely low wavelengths (such as 193 nm), the design of transparent polymers is a difficult problem due to the tendency for a wide variety of organic groups to absorb strongly in this ultraviolet wavelength range. Substituted polynorbornenes (PNB) are one potential material solution for providing transparent photoresist polymer resins for photolithography at 193 nm. The polynorbornene (PNB) backbone provides a relatively transparent alicyclic polymer structure with suitable plasma etch resistance. For a 193 nm photoresist resin, the characteristics of a pendant side group include: solubility in the common aqueous alkaline developers used for resist processing, ability to add acid-labile protecting groups to the polymer for producing chemically amplified resists resins with high contrasts, and reduction of the absorbance coefficient of the polymer due to the incorporation of fluorine. - Typically, hexafluoroisopropanol has been the pendant side group of choice in 157 nm lithographic photoresist design, though trifluoroisopropanol and tetrafluoroethane have been investigated as dilatants.
One of the most important characteristics of a polymer resin in terms of its suitability for use in photoresist applications is its dissolution behavior. Resist polymers must dissolve cleanly into an appropriate developer without forming a gel or swelling to any significant degree. It is also critical that resist resins can be produced that exhibit a controlled and reproducible dissolution rate in order to minimize batch-to-batch resist performance variation. Such control over polymer dissolution behavior can be achieved by methods such as blending various molecular weight polymer fractions together to achieve a composite resin with optimized performance. Therefore, in order to gain a better understanding of the basic polymer resin behavior and to aid in polymer design and resist formulation, a detailed investigation of the dissolution behavior of bis-trifluoromethyl carbinol substituted polynorbornene homopolymers (HFAPNB) has been performed in this work.
Figure 2: Dissolution Behavior of Substituted Polynorbornenes Specifically, reductions in the extent of hydrogen bonding in the HFAPNB polymer as molecular weights increase above 10,000 Mw were found to result in large changes in the polymer dissolution rate in aqueous base developers (see Figure 2).
Figure 3: Effect of Molecular
weight on Interchain hydrogen bonding for substituted polynorbornenes.
Polyhydroxystrene (PHOST, a common 248 nm resin) is shown on
this chart as a reference, due to its known ability to form interchain
hydrogen bonds.
Wide Angle X-Ray Diffraction studies showed that this reduction in hydrogen bonding with increasing molecular weight appears to be due to the disruption of intermolecular order in the polymer film (shown in Figure 4).
Figure 4: Wide Angle X-Ray
Diffraction Patterns for TFSPNB
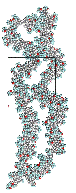
Based on molecular modeling results (see Figure 5), it is believed that reductions in intermolecular ordering in the polymer with increasing molecular weight are the result of increased numbers of “kinks” in the unusual secondary structure of the substituted polynorbornenes (see Figure 6). , , For the molecular modeling, there is an assumption that the pendant side group does not alter the properties of the substituted polynorbornene backbone.
Figure 5: Period Cell Diagrams for Low and High Molecular weight HFAPNB chains
Figure 6: Probability of formation of kinks in an HFAPNB polymer chain as a function of molecular weight (Mw).
While studying the dissolution behavior of HFAPNB, it was questioned whether the ability of the side group to form hydrogen bonds would alter the dissolution behavior of substituted polynorbornenes. Concerning dissolution studies, the only requirements of a side group are acidic hydrogen(s) for the acid-base reaction, and normal dissolution (i.e. no swelling, roughening, or formation of large interfacial gel layers) in aqueous base developers. After initial studies, the trifluorosulfonamide substituted polynorbornene exhibited normal dissolution (with minimal swelling) in aqueous base developers. From literature, it is known that the trifluorosulfonamide group has a higher propensity to form hydrogen bonds than the hexafluoroalcohol side group (see Figure 7). , Unlike HFAPNB, TFSPNB exhibits a dissolution behavior similar to most phenolic resins with a slope of -0.28 on a log-log plot (see Figure 2). The high dissolution rates of TFSPNB in aqueous base developer can be attributed to the low pKa of this polymer.
Figure 7: FTIR Spectrum for HFAPNB
and TFSPNB polymer films
In Figure 3, it is found that hydrogen bonding in TFSPNB polymers is nearly independent of molecular weight. There is a slight decrease in hydrogen bonding as molecular weights increase from 5 to 30,000 Mw, though this change is insignificant when compared to the disruption of interchain hydrogen bonding found in HFAPNB. This would suggest that the changes in the secondary structure are masked by the extensive interchain hydrogen bonding between TFS groups, which leads to “normal” dissolution behavior in aqeouse base developers.
References:
|




 (a)
(a)